如何从0到1搭建医疗器械质量管理体系:超全指南+避坑攻略,轻松拿证!【建议收藏】
质量管理体系漏洞的代价有多高?
《2021-2025 年医疗器械召回统计分析报告》显示,质量体系缺陷被召回的产品数量惊人。
2025年4月首家因质量管理体系严重缺失被责令停产的企业,详见《质量管理体系成摆设?三瑞事件警示:5大漏洞直接触发停产!》
质量管理体系如此重要,但从 0 到 1 搭建体系,要应对复杂法规、规划硬件设施、构建文件系统…… 千头万绪,不知从何下手?
别慌!这篇文章就为你呈上一份超详细的 “搭建指南”,带你一步步搭建起坚不可摧的质量管理体系。
不仅适用医疗器械生产企业,也适用于医疗器械经营企业。
前期策划:打好地基,才能盖高楼
1、法规与标准:找到你的指南针,法规是质量管理体系的根基。
企业生产(经营)产品不同,对应的法规有差异。
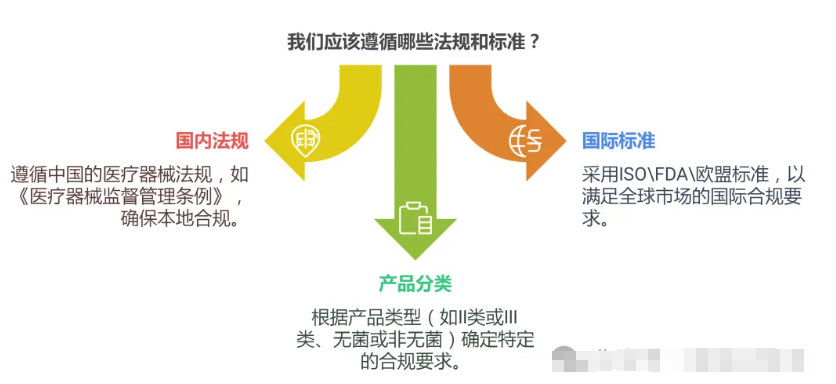
这些法规就像一张张地图,指引企业走向合规的彼岸。
别小看这一步,它决定了企业未来的方向。
根据企业实际情况,梳理出适用的法规和标准,组织人员学习、消化吸收,为质量管理体系的建立和运行提供制度保障。
2、硬件规划:给企业一个舒适的家
厂房和设施是企业的骨架。
洁净区、无菌区、仓储区,每一处都需要根据产品特性量身定制。
设备呢?同样需要根据产品特性量身采购。
买哪些设备?要自动还是手动?要大设备还是小设备?
企业规模和条件不同,要求自然也不一样。
结合企业实际情况,通过详细的市场调研、比较,列出详细的设备采购清单,标注好具体的要求,既能确保设备采购及时到位,又为后续文件顺利编写提供保障。
3、风险管理:未雨绸缪,才能防患于未然
风险无处不在。产品风险管理贯穿产品全生命周期。
用FMEA(失效模式与影响分析)等识别产品全生命周期的风险。
对于关键工序,比如灭菌、无菌灌装,更需要制定严格的措施,确保万无一失。
4、设计组织架构:让每个人都知道自己的位置
企业的组织架构就像一个精密的齿轮系统。
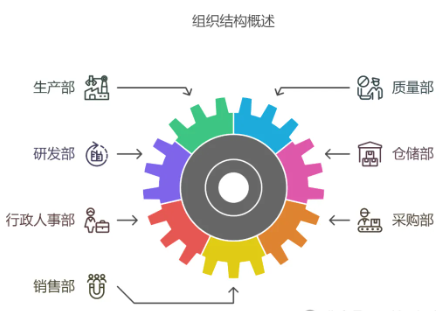
质量管理机构是核心,生产部、质量部、研发部、仓储部,每个部门都有自己的职责。
关键岗位人员更是重中之重——企业负责人是掌舵人,管理者代表是体系的守护者,质量负责人是监督员,生产负责人是流程的管理者,检验员是质量的守门员。
不同部门,不同岗位的人员在一切相互配合,才能让“精密的齿轮系统”正常运转。
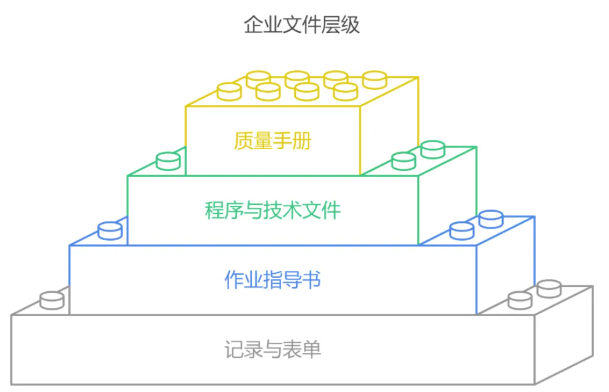
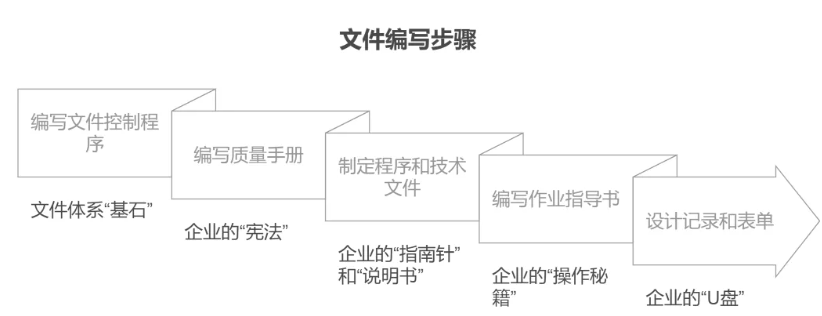
文件体系:让规则成为企业的语言
一级文件:质量手册
质量手册是企业的“宪法”,它告诉你企业的质量方针、目标、体系范围、组织架构和引用标准。
比如,“以客户为中心,提供零缺陷产品”就是一句简单而有力的方针。
二级文件:程序文件与技术文件
程序文件是企业的“指南针”,告诉你“做什么(what)”和“为什么做(why)”。包括文件控制程序、记录管理程序、设计开发控制程序等。
技术文件则是企业的“技术说明书”,是产品、物料、设备等要达到什么样的标准。涵盖质量标准、生产工艺规程、验证方案等。
三级文件:作业指导书(SOP)
SOP是企业的“操作秘籍”,它告诉你“如何做(how)”。
无菌操作SOP、设备操作SOP、检验方法SOP,每一份都至关重要。
四级文件:记录与表单
记录是企业的“U盘”,它确保每一步都有迹可循。
生产记录、检验记录、设备维护记录,这些看似枯燥的表格,其实是企业合规的基石。
根据企业的实际情况,详细梳理出每一层级所对应的文件,就形成了体系文件目录,按照文件目录分配编写任务,进度和质量都有保证。
2、文件编写:从规则到行动
它就像文件体系的“基石”,确保所有文件的编制、审核、批准、修订和作废都井然有序。
第二步,编写质量手册
明确企业的质量方针和目标。
《文件控制程序》和质量手册编写好后,一定要对文件编写人员进行系统培训,确保每一位文件编写人员都正确理解,并能有效执行。
第三步,制定程序文件和技术文件
程序文件要覆盖所有核心流程,比如采购、生产、检验。
技术文件包括标准、生产工艺、验证和确认等文件。
第四步,编制作业指导书
作业指导书在编写过程中,要遵循二个原则:
(1)相关内容要与质量手册、程序文件和技术文件保持一致。
(2)尽可能细化操作步骤,明确要求和注意事项。
第五步,设计记录与表单
也要遵循二个原则:
(1)要与质量手册、程序文件和技术文件、作业指导书保持一致。
(2)确保与产品质量有关的每一步都有记录支持。
体系实施:让规则落地生根
1、培训与意识:让每个人都成为合规的践行者
质量管理体系文件、法规要求,这些内容必须深入人心。
特别是关键岗位,比如管理者代表,必须通过ISO 13485内审员培训。
定期强调合规性、风险意识和客户导向,让每个人都能感受到质量的重要性。
从企业创建初期就建立“全员参与”的意识。
2、试运行:让体系在实践中成长
模拟生产是试运行的核心。
选择典型产品,全流程试生产,既验证了体系的适用性,又能验证部门内和部门间配合问题。
对发现的问题,及时寻找原因,制定解决办法。
切记,一定要将新获得的技巧和经验,通过修订文件的方式,让体系在实践中不断完善。
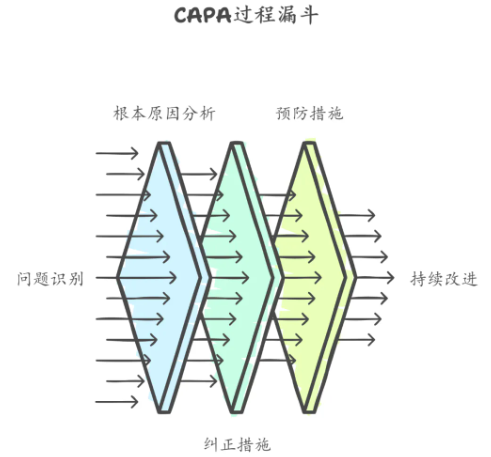
3、内部审核:发现问题,解决问题
内审是体系的“体检”。
每年至少一次,覆盖所有部门和过程。
发现问题是持续改进的前提。
通过制定纠正预防措施(CAPA),跟踪验证效果,确保问题彻底解决。
4、管理评审:让体系不断进化
管理评审是体系的“头脑风暴”。
每年至少一次,由最高管理者主持。
内审结果、客户投诉、法规变化、质量目标达成情况,这些都是评审的输入。最终输出的,是体系改进的方向和资源需求。
体系认证与准备:让合规成为企业的底气
1、申请生产许可证:准备充分,才能胸有成竹
资料准备是申请生产许可证的关键。
根据申报资料要求逐一准备,装订,申报。
正式检查前,一定要认真梳理《医疗器械生产质量管理规范》和相应的指导原则,把关键项梳理出来逐一核对,确保无遗漏。
2、迎接审核与整改:让问题成为改进的契机
审核是体系的“考试”,戏称“体考”。
检查过程中,快速响应检查员提出的任何问题和要求,第一时间提供证据,比如记录、现场演示等,是顺利通关的“秘籍”。
整改则是体系的“补课”。
组织人员,制定详细的整改方案,第一时间提交完整的整改报告,拿证就有了确切的保障!
持续改进:让体系永远在路上
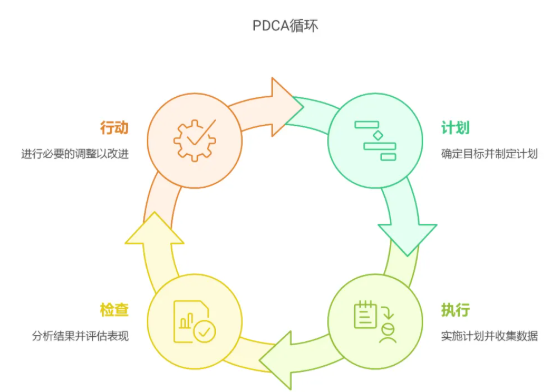
1、数据分析与监控:用数据说话
质量数据是体系的“眼睛”。
收集生产、检验、客户反馈等一切数据,分析趋势,比如不合格率、退货率等,洞察隐藏的质量信息。
再通过PDCA循环(计划-执行-检查-处理),不断优化流程,持续改进质量管理体系。
2、外部审核与认证:让第三方为你的体系背书
有条件的企业,可邀请认证机构进行ISO 13485或GMP认证审核,确保体系经得起考验。
3、文件动态更新:让规则与时俱进
切记:法规变化、产品更新、流程改进,这些都需要及时修订文件。
体系文件的版本号和修订记录,是确保全员使用最新版本的关键。
特殊要求:应对复杂场景的挑战
1、不良事件与召回管理:未雨绸缪,才能临危不乱不良事件监测是体系的“雷达”。
要建立报告系统,主动收集用户反馈、投诉和事故。
产品召回机制则是体系的“急救箱”。
要制定召回预案,明确分级、流程和责任部门。
2、委托生产与外包管理:把信任变成责任
供应商管理是体系的“外延”,是保障产品质量的源头。
必须进行资质审核、定期评估,通过质量协议和现场审计监督供应商表现。
委托生产要求则需要签订质量协议,定期审计受托方质量体系。
医疗器械质量管理体系不是冷冰冰的文件堆砌,而是企业生存的护城河。 从前期策划到持续改进,每一步都关乎产品安全、市场信任与品牌口碑。 始终相信:质量是企业的生命线,合规是企业的底气。 从今天开始,让合规成为你的底气,让质量成为你的骄傲!
 售前咨询专员
售前咨询专员